
Yang ZHANG
Determining structure and function of protein and RNA molecules is a cornerstone of modern biology and medicine. The Yang Zhang Lab develops novel bioinformatics algorithms, built on cutting-edge artificial intelligence (AI) technique and physics-based atomic force field, to accurately model and redesign the structure and function of proteins and RNAs. While one goal of the studies is to reveal the fundamental relationship between genome sequence and protein structure and function, the Zhang Lab is particularly interested in the extension of AI-based computational approaches to medicine design and drug discovery, with a focus on cancer therapeutics and treatment.
zhang[at]nus.edu.sg
Senior Principal Investigator, Cancer Science Institute of Singapore
Professor of Computer Science, School of Computing, NUS
Professor of Biochemistry, Yong Loo Lin School of Medicine, NUS
You may learn more about Prof Zhang here: https://zhanggroup.org/
The research of Prof. Yang Zhang Lab focuses on protein folding and structure prediction, protein design and engineering, and structure-based protein function annotation and drug discovery (Fig 1). Several methods developed in the Zhang Lab have been recognized as the world’s best. Among them, I-TASSER (as ‘Zhang-Server’ and ‘UM-TBM’) was ranked as the No 1 most accurate method for automated protein structure predictions in the last nine CASP experiments (CASP7-15 in 2006-2022), which are biennial community-wide blind contests designed to benchmark the state of the art of the field. Accordingly, the I-TASSER server (https://zhanggroup.org/I-TASSER/) becomes one of the most widely used platforms for protein structure and function predictions, serving more than 170,000 registered users from 160 countries.
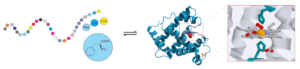
Figure 1. One principal goal of the research in Zhang Lab is to reveal the fundamental relationship between protein sequence, structure, and function.
In addition to I-TASSER, many of other tools developed in the Zhang Lab are widely used by the globe community and have generated important impacts on the community. For instance, TM-score (https://en.wikipedia.org/wiki/Template_modeling_score) and TM-align (https://zhanggroup.org/TM-align) programs are used as standard tools for protein structure analyses and selected as the default structure comparison methods by various systems including the Protein Data Bank (PDB) and the Debian Unix/Linux System. The idea of template profile-based protein structure prediction was later extended to protein design, where a novel evolution-based method, EvoDesign, was proposed to successfully design new protein domains and peptides to regulate cancer cell apoptosis pathway and to block the association of SARS-CoV-2 virus with human host cells, respectively.
Most recently, the Zhang Lab aspires to develop new artificial intelligence (AI) and deep learning methods to improve the scope and accuracy of protein folding and protein design, which represent two fundamentally important and inverse procedures of biological process (Fig 2). The Zhang Lab is among the first a few laboratories who have initiated the studies on deep machine-learning based protein and RNA structure predictions, while the artificial intelligence (AI) technique has eventually brought about revolutionary impact on the field of structural biology and life science. The Zhang Lab is particularly interested in the application of the AI-based computational algorithms for effective cancer therapeutics and treatment, with focuses on anti-cancer peptide-drug conjugates (PDCs), T-cell receptor (TCR)-based adoptive therapy, and antibody discovery and optimization.
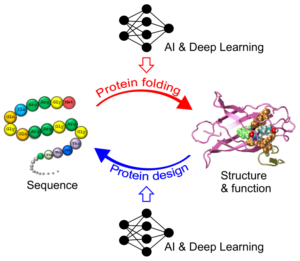
Figure 2. The integrations of AI and deep neural-network learning techniques significantly improve the power and accuracy of two inverse processes of protein folding and protein design.
Lab Members



