
Sriram SRIDHARAN
Cancer therapy designed to kill cancer cells, work by inducing DNA double strand breaks (DSBs). These DSBs overwhelm the cells forcing them to die. Highly conserved DNA repair mechanisms protect the integrity of the genome by repairing these damages. Recently, DNA repair inhibition has emerged as a promising strategy for personalized therapy. Furthermore, employing DNA-damaging agents in combination with drugs that inhibit DNA repair pathways can have synergistic effect on eliminating cancer cells. DSBs are also introduced in the genome as part of normal physiological processes such as antibody diversification, meiotic recombination. These DSBs can sometimes get repaired in an erroneous fashion leading to human diseases including cancer. Thus, DSBs have a very important functions in physiological processes, disease initiation as well as in therapy. Studying DSBs- how they are induced, how they are repaired and how they impact the cell – is critical to have a mechanistic understanding of many fundamental processes.
srirvs[at]nus.edu.sg
Special Fellow, Cancer Science Institute of Singapore
–
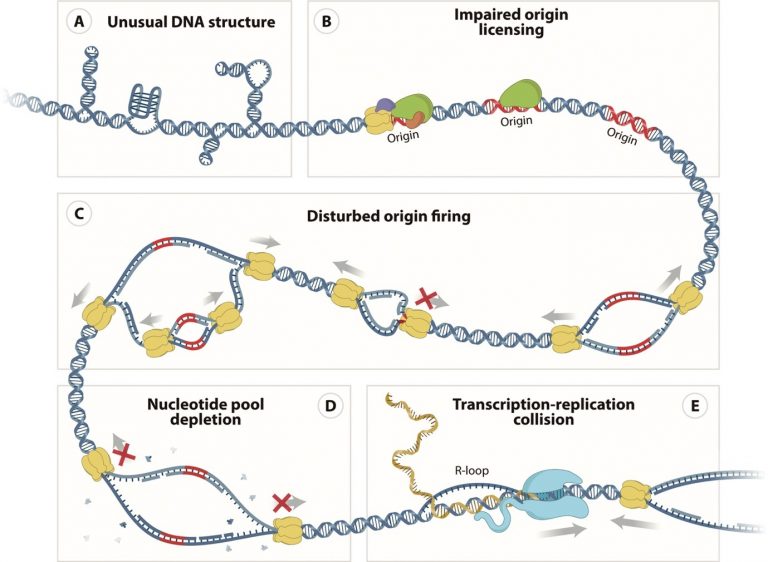
Fig. 1: Sources of replication fork stalling and replication stress. Such situations can give arise to duplicated DNA with errors which gives arise to genome instability.
Cells undergoing cellular replication need to duplicate their DNA, a complex process regulated spatially and temporally by a plethora of proteins. Dysregulation of this critical process results in induction of genome instability, a hallmark of cancer. Consequently, any action that interferes with the ability of the cell to faithfully replicate their DNA is known as replication stress and can induce genome instability, an all-encompassing term that reflects myriad forms of DNA damage and the ensuing adverse consequences that come from an inability to repair and process this damage. Therefore, understanding the genomic contexts in which replication stress is induced is crucial to the understanding of cancer etiology.
Replication stress either through endogenous or exogenous sources can cause incomplete and unfaithful replication by stalling and/or terminating the replication machinery prematurely resulting in DNA lesions, the most deleterious being the Double Strand Breaks (DSBs). There are many impediments to progression of replication forks as shown in Fig. 1. When cells are subject to replication stress, replication forks are stalled and triggers the activation of downstream process that promotes resolution of the stress and subsequent restart of the replication forks (RFs). These processes are collectively known as the S-phase checkpoint. This leads to DNA replication forks that are stalled to form various structures under a mechanism known as fork protection. The persistent stalling of RFs is characterized by stretches of single stranded DNA (ssDNA) which result from the uncoupling of the DNA helicase, which is used to unwind DNA ahead of the DNA polymerase and the DNA helicase itself. Stalled forks that are unable to restart resolve into DSBs which the cell tries to repair. DSBs can be repaired through two major pathways- homologous recombination (HR) and non- homologous end joining (NHEJ). Cancer therapeutics that target the DNA repair pathway, operate by elevating the level of endogenous replication stress in cancer cells and force them to undergo apoptosis. These drugs induce/elevate replication stress in various ways including nucleotide (dNTP) depletion, DNA polymerase inhibition, and inhibiting helicases necessary for restarting stalled replication forks.
Our overarching goal is to understand why, where and what of the (epi)genome acts as an impediment to faithful DNA replication. To this end, we will develop computational/statistical models that tease out the (epi)genomic contexts that make them barriers to DNA replication, response of the cells to this stress and mechanisms by which the cell overcomes this stress.
*indicates joint first author
Lab Members

